 |
|
|
|
|
 Back to 2001 2nd Quarter Table of Contents
Back to 2001 2nd Quarter Table of Contents
Abstract Relationship between depression of early protection against influenza virus infection and the decrease in the number of peripheral polymorphonuclear leukocytes (PMN) in cyclophosphamide (CY)-treated mice was investigated using d-Lenolate (Lenolate), which had been shown to exert a potent restorative effect on leukocytopenia in immuno-compromised hosts. Following intranasal inoculation with influenza virus (2.0 x 103 PFU) into untreated mice, the pulmonary virus titer progressively increased during 3 days and decreased gradually from the day 7 after infection. The treatment of mice with CY 2 days before infection markedly enhanced the pulmonary virus multiplication from the early phase of infection, and the higher virus titer was maintained thereafter. When mice were given Lenolate after CY-treatment, virus titers from the early to late phases of infection were lower than those in CY-treated controls not given Lenolate. The number of peripheral PMN in CY-treated mice rapidly decreased and returned to normal levels only 9 days after the treatment, while such leukocytopenia was prevented to some extent and the leukocyte count was restored completely up to 7 days by post-CY-treatment with Lenolate. Furthermore, the treat
ment with Lenolate augmented the inactivation of virus by PMN. However, the virus inactivation by alveolar macrophages was modified only slightly by Lenolate treatment. In addition, Lenolate-treatment could potentiate antibody-dependent cell-mediated cytotoxicity of PMN against the virus-infected target cells, and the production of serum neutralizing antibody to influenza virus in untreated and CY-treated mice. Lenolate revealed neither antiviral nor interferon inducing-activities. The augmented protection against influenza virus infection in immuno-compromised hosts by Lenolate may be attributed mainly to a partial prevention of a decrease in the number and activity of PMN and a recovery from the depressed values. Key Words: d-Lenolate, Influenza virus, PMNs, CY Introduction The significance of phagocytes consisting mainly of polymorphonuclear leukocytes (PMN) and macrophages has been investigated in terms of the relative contribution to early protection against bacterial infections using their differential susceptibility to X-irradiation and carrageenan.1,2,3 PMN, X-ray-sensitive and carrageenan-resistant, contribute mainly to the early phase of protection against extracellular bacteria, such as Pseudomonas aeruginosa (P. aeruginosa),2 Escherichia coli (E. coli )3 and Streptococcus pneumoniae (S. pneumoniae),4 while the protection against intracellular bacteria such as Listeria monocytogenes (L. monocytogenes)1,2 highly depends on carrageenan-sensitive macrophages. Among macrophage series, tissue-fixed macrophages are X-ray-resistant and blood monocytes are X-ray-sensitive. In contrast, the relative contribution to the early protection against virus infections has not been well investigated. Especially, a few studies are available on a protective role of PMN in the early phase of virus infection. Our preceding study demonstrated that PMN played a protective role in the early phase of intranasal infection with low (2.0 x 103 PFU) and high (2.0 x 104 PFU) doses of influenza virus, whereas alveolar macrophages contributed to the early protection.5 On the other hand, it has been shown that the decrease in the number of PMN in the peripheral blood correlated with the risk of bacterial infections6,7 and also that the restoration from leukocytopenia prevented opportunistic infections induced by the treatment with anticancer drugs.8-11 Furthermore, inoculation of several different types of viruses resulted in severe infections in hosts compromised by X-irradiation12,13 or cyclophosphamide.14,15,16 However, there is little evidence concerning the correlation between restoration of PMN numbers and reconstitution of early protection against viral infection in the immunocompromised state. D-Lenolate (Lenolate) derived from extract of olive leaves, has been found to be effective, in vitro, against Salmonella enteritidis and enterotoxin B.17 A recent study in our laboratory indicated that Lenolate, exerted effects on the restoration from leukocytopenia resulting in augmented protection against several bacterial infections in immunocompromised mice.13 In this study, we investigated the effect of Lenolate on early protection against influenza virus infection and number of PMN in immunocompromised hosts. Materials and Methods Animals. Female BALB/c mice were purchased from Japan Charles River (Tokyo) and used at 8 weeks of age. Drugs. d-Lenolate (Lenolate), a patented sterio extraction from olive leaves, was obtained from East Park Research (Henderson, NV, USA). In this study, prospective recipients of influenza virus challenge (day 0) received oral injections of 500mg/kg of Lenolate daily for 7 days before the influenza infection and functions or phenotypes of PMNs were assayed. Cyclophosphamide (CY; Endoxan, Shionogi & Co., Ltd., Osaka) was dissolved in sterile distilled water (20 mg/ml) before use. Mice were intraperitoneally injected with CY (200 mg/kg) 2 days before virus infection. Antisera. Rabbit anti-asialo GM1 antiserum was obtained from Dr. K. Okumura, Juntendo University (Tokyo). In the experiments performed for this study, the mice were injected intravenously with 20 μl anti-asialo GM1 six times at 2-day intervals, beginning 1 day before the virus inoculation.18 Intravenous infection of anti-asialo GM1 antiserum completely abolished NK activity against lymphoma cell line (YAC-1) in spleen cells from BALB/c mice. Virus. Influenza virus strain A/PR/8/ 34 (H1N1) obtained from the Japan National Institute of Health (Tokyo) was passed twice in the allantoic cavity of 10-day-old embryonated chicken eggs for 48 h and stored as infectious allantoic fluid at -70ºC before use. The virus titer was 5.0 x 108 plaque forming units (PFU/ml) by a plaque assay with Mardin Darby Canine Kidney (MDCK) cells. Mice were infected intranasally by a head exposure chamber (Sibata Scientific Technology Ltd., Tokyo). Cell lines. MDCK cells and L cells were cultured in Eagle’s minimal essential medium (MEM; Grand Island Biological Co., Grand Island, N.Y.). Preparation of anti-influenza virus antiserum. Mice were intravenously inoculated with 1.0 x 108 PFU of the virus and boosted with the same virus 1 week after priming. One week later, mice were anaesthetized and blood was collected from the axially artery, the sera were obtained by centrifugation. The titer of neutralizing antibody was 1:1024 in plaque assay. Virus by a MDCK cell plaque assay. A plaque assay was carried out by a small modification of the method of Luckacher et al.19 Lungs were removed from mice after virus infection and homogenized manually with PBS to give a 10% homogenate. The homogenate was dispersed by sonication (200W, 2A for 10 min) and centrifuged (10,000g, 2 min) to remove coarse debris, and stored at -70ºC until titration. Tenfold dilutions of the supernatant were prepared in PBS supplemented with 0.25% bovine serum albumin (BSA; Fraction V powder, Sigma, St. Louis). MDCK cell monolayers prepared by seeding 4.0 x 105 cells in 2 ml MEM supplemented with 10% fetal calf serum (FCS; GIBCO) into tissue culture dishes (35 x 10 mm) (Falcon 1008; Becton Dickinson Labware, Lincoln Park, New Jersey) 2 days before were infected with 0.1 ml of the virus dilution for 60 min at 25ºC. After virus adsorption, the virus suspension was discarded and each monolayer was washed twice with 1.5 ml of Hanks’ solution (GIBCO) to remove trypsin inhibitors in the organ homogenate, and then over-laid with 2 ml of agar medium containing 0.45% BSA, 2 mM glutamine, 100 mg/ml of glucose, 2 g/ml of folic acid, 2 μg/ml of biotin, 100 μg/ml of DEAE-dextran, 100 μg/ ml of streptomycin, 100 U/ml of penicillin, 0.85% purified agar (Difco, Detroit) and 2.5 μg/ml of trypsin (Chemical Company, St. Louis). After incubation for 2 to 3 days in a 5% CO2 atmosphere at 37°C, plaques were counted. Each titration point of individual homogenates was assessed in triplicate cultures. Results were expressed as the mean PFU±SD per organ from 8 mice. Serological testing. Neutralizing anti-bodies were determined by plaque assay in MDCK cells. Sera obtained from 8 mice infected with the virus were pooled and treated with a receptor destroying enzyme (RDE; Takeda Pharmaceutical Co., Ltd., Osaka) for 18 h, and then the treated sera were inactivated at 56ºC for 30 min. Two-fold dilutions of the sera were prepared in PBS supplemented with 0.25% BSA and mixed with an equal volume of the virus suspension (1,000-2,000 PFU/ml), and the mixture was incubated at 37ºC for 1 h. The mixture (0.1 ml) was plated onto MDCK cell monolayers and neutralizing dose 50 (ND50) of the sera was determined by calculating from reciprocal dilution to reduce 50% of the virus plaque number in the virus dilution plus normal mouse serum. Interferon (IFN) titer in the sera was determined by the method of Neumann and Sorg.20 Briefly, L cells (2,000 cells/ l) were plated into each well of microplates (Falcon 3034) and incubated at 37ºC for 6 h; two-fold dilutions of the sera (15 μl) from eight mice were added into the well after discarding the culture supernatant, washed twice with MEM and vesicular stomatitis virus (VSV, New Jersey strain; 50 TCID50) in 10 μl aliquots was added to each well. The IFN titer was assessed by the endpoint showing 50% reduction of the cytopathic effect. Preparation of PMN and alveolar macrophages. PMN were collected from the peritoneal cavity of mice given 1 ml of glycogen (1.0% in PBS) 3 h before. Peritoneal exudate cells were harvested in Hanks’ solution supplemented with 0.5 units of heparin, washed with RPMI 1640 medium (GIBCO) supplemented with 2 mM glutamine, 100 μg/ml of streptomycin, 100 U/ml of penicillin and 10% of FCS (supplemented RPMI 1640), and PMN were purified by centrifugation (300 g, 30 min) after layering onto Monopoly Resolving Medium (Flow Laboratories, Inc., Versinia). The PMN-enriched fraction was collected and washed three times to remove the medium, and resuspended in the supplemented RPMI 1640. Alveolar macrophages were collected by washing the lungs of mice. The trachea was cannulated with a needle and anchored by suturing. The lung was washed with PBS including 0.02% EDTA (10 ml) and cells were collected by centrifugation; then, resuspended in supplemented RPMI 1640. The purity of PMN and alveolar macrophage suspensions was over 98%, determined by histological observation after Giemsa staining. Virus infection to phagocytes in vitro. PMN or alveolar macrophages (2.5 x 105 cells) were infected with influenza virus at multiplicity of infection (m.o.i.) of 0.1, 1.0 or 10 PFU/cell in a volume of 0.2 ml at 25ºC for 2 h. Cells were washed 5 times with MEM, suspended in the supplemented RPMI 1640 and cultured into 24 well-multidishes (Falcon 3047) at 37ºC for 24 or 48 h in a 5% CO2 atmosphere. Infected cells were washed 4 times with MEM, resuspended in 1 ml of PBS supplemented with 0.25% BSA and disrupted by sonication for 5 min (200W, 2A). The virus titer in the infected cells was determined by plaque assay in MDCK cell and the results were expressed as the mean PFU ±S.D. per 106 cells of triplicate cultures. Assessment of an inhibitory effect on virus replication by phagocytes in vitro. MDCK cells (1.0 x 105 cells/ml), seeded into 24 well-multidishes (Falcon 3047) 18 h previously, were infected with influenza virus at an m.o.i. of 0.1, 1.0 or 10 at 25ºC, and incubated in a 5% CO2 atmosphere at 37ºC for 6 h. One ml of suspensions of PMN or alveolar macrophages were added to the MDCK cell cultures in the presence or absence of heat-inactivated mouse anti-influenza antiserum or normal mouse serum (NMS) (1:200), respectively, and then cells were cultured at 37ºC in a 5% CO2 atmosphere for 24 or 48 hours. Cells and supernatants were harvested after one frozen and thawing cycle, and virus titers in the cultures were determined by MDCK cell plaque assay. Results were expressed as the mean PFU±SD per 105 cells of triplicate cultures. Measurement of direct cytotoxic and antibody-dependent cell-mediated cytotoxic (ADCC) activities by phagocytes. MDCK cells (1.0 x 104 cells/well), seeded into flat bottomed 96 well-microtiter plates (Falcon 3072) 18 hours before, were incubated with 1.11 MBq of Na251CrO4 at 37ºC for one hour, washed four times with MEM, and subsequently cultured for three hours. Cells were infected with influenza virus at anm.o.i. of 10 PFU/cell at 25ºC for one hour, washed twice with MEM and cultured in the supplemented RPMI 1640 for six hours. Phagocytes (5.0 x 105 cells/well) were added to the cell cultures in the presence or absence of anti-influenza serum or NMS (1:200), respectively, and subsequently incubated for 6 to 18 hours. Radioactivity in the culture supernatant was determined by an autogamma counter and cytotoxic or ADCC activities of phagocytes were calculated as percentage of 51Cr released from following formulas: Cytotoxic activity (%)= release by phagocyte - spontaneous release X100 total counts - spontaneous release ADCC activity (%)= release by phagocyte - spontaneous release X100 total counts - spontaneous release Spontaneous release from target cells infected with the virus alone usually ranged from 3 to 5% of total counts. Total counts were determined by target cell lysis after freezing and thawing the culture with supernatant. Determination of antiviral activity in vitro. MDCK cells (1.0 x 105 cells/ml) were seeded into 24 well-multidishes (Falcon 3047) 18 h before use and influenza virus was infected at an m.o.i. of 1.0. Lenolate was added to the wells two hours after incubation in 5% CO2 atmosphere at 37ºC and cells were further cultured for 18 h. Cultured cells and supernatants were harvested after one frozen and thawing cycle, sonicated for 5 minutes (200W, 2A) and centrifuged (3,000 rpm) at 4ºC for 10 minutes. The virus titer in the supernatant was determined by MDCK cell plaque assay and results were expressed as the mean PFU±SD per 105 cells of triplicate cultures. Histological observations. Blood specimens were collected by puncture of the retroorbital venous plexus. Total numbers of leukocytes in the blood and peritoneal exudate were counted after staining with Turk’s solution and differential counts were carried out after staining the smears with Giemsa solution. Results Mortality rate of mice after influenza virus infection. Intranasal infection with 2.0 x 104 PFU of influenza virus resulted in a mortality rate of 100% in CY-treated mice but no death in untreated and Lenolate-treated groups of mice within 15 days after the infection (Figure 1, p. 6). The mortality rate of mice treated with Lenolate after CY-pretreatment decreased to 30% (Figure 1, p. 6). Mice of all groups infected with 2.0 x 103 PFU of influenza virus survived beyond 15 days in experiments performed under the same experimental conditions shown in Figure 2,4, p. 7,9 Virus titer in the lung of mice infected with influenza virus. The virus titer in the lung of normal mice was 3 x 102 PFU at 1 h after infection. The titer increased progressively during five days, then it decreased gradually from day seven and reached an undetectable level by day 13 (Figure 2, p. 7). When CY-treated mice were used as hosts, the virus titer at one hour post infection was not significantly different from that in normal mice. The titer increased rapidly up to day three after infection, reached a maximum on days seven to nine and was very high by day 15, the last time virus titration was carried out. The titers in CY-treated mice were significantly higher than those in untreated mice from day one to day 15 (Figure 2, p. 7). When mice were given Lenolate after CY-pretreatment, the pulmonary virus titers on days one to five decreased to levels similar to those of normal mice. The virus titers in CY-treated and Lenolate-given mice decreased gradually from day seven but the levels on days seven to nine were higher than those of normal mice (Figure 2, p. 7). Time-course observation of virus titers in the lung of normal mice given Lenolate showed a pattern similar to that in normal mice but the level was significantly lower than that in normal mice during the period from day one to day 13 (Figure 2, p. 7). Enhanced restoration of decreased leukocyte count in the peripheral blood by Lenolate. When mice were treated with CY, the number of peripheral PMN decreased rapidly and reached a minimum level at three days after CY-treatment. The PMN numbers gradually increased from day 7 and was completely restored by day 11 (Figure 3, p. 8). In mice given Lenolate one and two days after CY-treatment, the number of leukocytes decreased during three days after CY-treatment and started to recover from day three, and then reached a completely normal level by day seven (Figure 3, p. 8). The recovery of leukocyte number occurred several days before the elimination of virus in CY-treated and Lenolate-treated mice. This interval may be needed for the maturation and/or activation of PMN. Antibody titer to influenza virus after intranasal infection. Antibody was not detectable up to day three after virus infection in any groups of mice (Figure 4, p.9). The antibodies were detected on day five in mice treated only with Lenolate and on day seven in untreated mice, the titers increased rapidly thereafter in both groups of mice (Figure 4, p. 9). The titer in CY-treated mice gradually increased from day 11 and reached approximately 1/10 that of untreated mice on day 15. Lenolate treatment could potentiate the antibody production from day nine after virus infection in such CY-treated mice (Figure 4, p. 9). Viral multiplication in influenza virus-infected phagocytes. The multiplication of virus in PMN and alveolar macrophages obtained from Lenolate-treated mice was compared with that of the virus in phagocytes from normal mice. When PMN of Lenolate-treated mice were infected with Figure 1. Mortality rate of BALB/c mice after intranasal inoculation with 2.0 x 104 PFU of influenza virus.
100 50 0 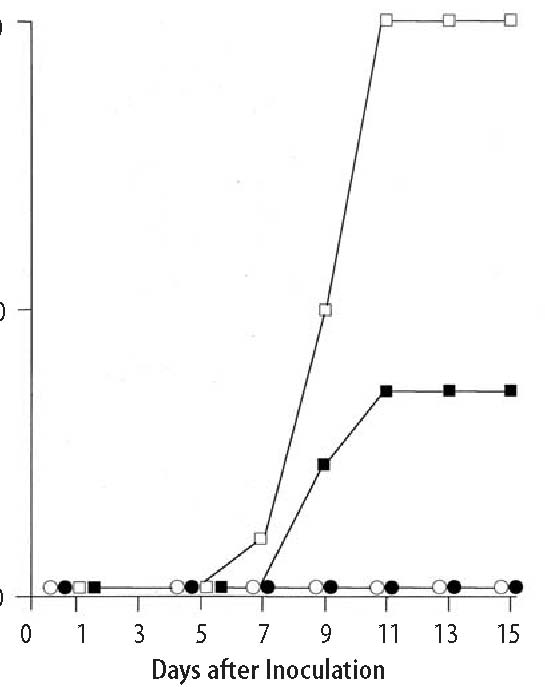 influenza virus at an m.o.i. of 1.0 or 10.0 PFU/cell, the virus titer in the PMN in culture was reduced more markedly compared to that in normal PMN (Table 1, p. 8). In contrast, the virus multiplication in alveolar macrophages was suppressed only slightly by Lenolate-treatment (Table 1, p.8). These results suggest that the qualitative effect of Lenolate ascribes mainly to the activation of PMN but only partially to the activation of alveolar macrophages in addition to its quantitative effect, namely, the enhanced recovery of leukocyte numbers. Inhibition of virus replication by PMN in vitro. When normal PMN were added to virus-infected MDCK cell culture, the virus titer was reduced to approximately 1/5 to 1/ 10 that of the culture without PMN 24 or 48 hours after virus infection (Table 2). The addition of PMN from Lenolate-treated mice inhibited the virus replication more markedly than that of normal PMN (Table 2, p. 9). Cytotoxic and ADCC activities of phagocytes. After PMN and alveolar macro-phage suspensions obtained from normal mice were added to the virus-infected MDCK cell cultures without anti-influenza serum, PMN and alveolar macrophages exhibited a weak cytotoxic activity against virus-infected MDCK cells (Table 3, p.11). The addition of anti-influenza serum to the culture in the presence of PMN, facilitated the cytotoxic activity of PMN, more effectively than the addition of NMS (Table 3, p. 11). The treatment Figure 2. Enhancement by cyclophosphamide pretreatment of pulmonary virus multiplicity in BALB/c mice inoculated intranasally with 2.0 x 103 PFU of influenza virus and its inhibition by Lenolate treatment.
 7 6 5 4 3 2 1 0 with Lenolate augmented the ADCC activity of PMN but not the direct cytotoxicity against virus infected MDCK cells (Table 3, p.11). In contrast, the cytotoxic activity and ADCC activity of alveolar macrophages were not augmented by Lenolate-treatment. Antiviral activity of Lenolate in vitro. Lenolate (0.01-100 ng/ml) did not show any antiviral activity in in vitro cultures infected with influenza virus (data not shown). IFN levels in the serum. In those experiments, serum IFN was not detectable in any group of either untreated, CY-treated, Lenolate-treated or CY plus Lenolate-treated mice (data not shown). Effect of NK cell depletion on influenza virus infection. To ascertain whether or not the NK cells participate in the protection of mice against virus infection, intact and NK cell-depleted mice (mice injected with anti-asialo GM1) were inoculated intranasally with 2.0 x 104 PFU of influenza virus, and the survival rate was determined. There was no significant difference of survival time between the mice receiving multiple injections of anti-asialo GM1 from day -1 and the mice receiving normal rabbit serum (NRS) (Figure 5, above). Further-Figure 3. Preventive and restorative effects of Lenolate on PMN level in the peripheral blood of cyclophosphamide treated BALB/c mice. 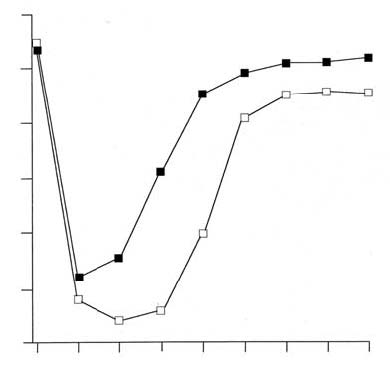 7 6 5 4 3 2 1 0 Table 1. Effect of Lenolate on viral multiplication in phagocytes infected with influenza virus) Drugsa Infected cellsb m.o.i. PFU x 10-2/106 cellsc 24 48h None PMN 1.0 14.0 ±1.5 1.5 ±0.2 Lenolate PMN 1.0 2.0 ±0.1 0.1 ±0.1 None Aveolar MØ 1.0 90.5 ±5.0 30.0 ±5.2 Lenolate Alveolar MØ 1.0 82.0 ±3.5 26.5 ±6.0 None PMN 10.0 230.0 ±32.2 35.2 ±8.0 Lenolate PMN 10.0 64.5 ±10.2 5.2 ±0.1 None Alveolar MØ 10.0 1025.4 ±202.6 582.4 ±95.0 Lenolate Alveolar MØ 10.0 995.2 ±212.0 410.0 ±66.4 a) Polymorphonuclear leukocytes (PMN) or alveolar macrophages (MØ) were prepared from normal mice or miceinjected per os with Lenolate (500 mg/kg) seven days previously.b) Cells (2.5 x 105) were infected with influenza virus at a m.o.i. of 1.0 or 10.0 at 25ºC for two hours.c) Virus titers in the cells were measured by MDCK cell plaque assay 24 and 48 hours after the injection. Resultswere expressed at the mean value ±SD in triplicate cultures. Figure 4. Production of neutralizing antibody in BALB/c mice infected intranasally with 2.0 x 103 PFU of influenza virus.
>1000 -1000 100 10 <8 0 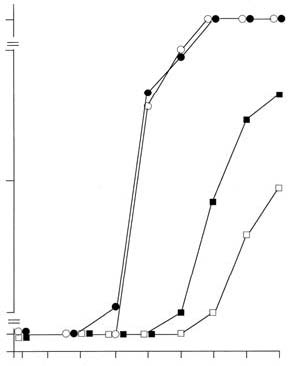 Table 2. Effect of Lenolate on the inhibition of virus replication by PMN in vitro. Effector Lenolate E/T m.o.i. PFU x 10-7/105 cellsd cellsa treatmentb ratioc 24 h 48 h None – 0.1 285.5 ±40.0 465.0 ±42.6 PMN – 10 0.1 40.6 ±8.4 27.2 ±4.0 PMN + 10 0.1 8.6 ± 0.4 2.0 ±0.5 None – – 1.0 750.0 ±90.5 492.0 ±80.0 PMN – 10 1.0 142.4 ±10.0 44.4 ±8.6 PMN + 10 1.0 35.2 ±1.0 8.0 ±0.8 a) PMN were added to MDCK cell cultures infected with influenza virus at an m.o.i. of 0.1 or 1.0, 6 h after the infection.b) Mice were injected per os with Lenolate (500 mg/kg) 7 days before obtaining the PMN. c)PMN were added to virus-infected MDCK cells at an effector to target cell (E/T) ratio of 10.d) Virus titers in the cultures were measured by MDCK cell plaque assay 24 and 48 h after the infection. Results wereexpressed as the mean ±SD in triplicate cultures. Figure 5. Effect of anti-asialo GM1 injection on survival of influenza virus-infected BALB/c mice. Mice which received 20 μl anti-asialo GM1 injection or 20 μl NRS according to the indicated schedule were inoculated intranasally with 2.0 x 104 PFU influenza virus on day 0. Each group was composed of 20 mice.  Days after virus inoculation NRS (
more, the mice injected with the antisera beginning from day 1 survived as long as the control mice. Discussion The data presented demonstrate that the early protection against influenza virus infection is depressed due to a decrease in the peripheral PMN level in immuno-compromised hosts and that the PMN level could be normalized. The prevention of decrease or rapid restoration of PMN level by Lenolate-treatment was primarily along with the restoration of depression induced with CY-treatment in the early protection against the intranasal infection with a low dose (Figures 1-3, p.6-8). It was already indicated that Lenolate had potential preventive and restorative effects on decreases in the number of peripheral PMN and bone marrow cells induced by anticancer agents; and, also that both phagocytic and bactericidal functions of PMN and macrophages were activated by the treatment with Lenolate (unpublished data). In the present study, Lenolate-treatment contributed to the inactivation of virus in PMN and facilitated the inhibition of virus replication by PMN in MDCK-infected cells cultures (Table 1,2, p.8,9). These results, therefore, strongly suggest that Lenolate stimulates PMN activity in addition to its ability to restore PMN number and could also contribute to early protection against the virus infection. However, the virus inactivation in infected alveolar macrophages was enhanced only slightly by the Lenolate treatment (Table 1, p.8), indicating that the restoration of early protection induced by Lenolate was not attributable to the activation of alveolar macrophages. In fact, the treatment of normal mice with Lenolate could diminish the pulmonary virus titer in the early stages when a low dose of virus (2.0 x 103 PFU) was used (Figure 2, p.7), alveolar macrophages do not contribute to the early protection.5 Furthermore, in our previous studies, intraperitoneal administration of carrageenan before intranasal infection with the high dose of influenza virus did not increase virus titre in the lung at the early phase of infection.5 These findings seemed likely to support the above interpretation. It has been reported that NK cells participate in non-specific resistance in several virus infections.21,22 In our study, intravenous injections of anti-asialo GM1 antiserum before or after the virus inoculation did not result in the increase of the pulmonary virus titre (Figure 5, p.10). Thus, it seems unlikely that NK cells contribute greatly to early protection against intranasal infection with influenza virus. Rodgers & Mims23,24 reported that murine alveolar macrophages might be capable of spreading the infection in vivo, since such infected macrophages were able to act as infectious centers for tissue culture cells. However, the present study indicated that both PMN and alveolar macrophages participated in early protection after intranasal infection with a high dose of the virus. After infection with a low dose, no contribution of alveolar macrophages to early protection could be demonstrated. ADCC but not nonspecific cytotoxicity of PMN against the virus in- Table 3. Effect of Lenolate on the cytotoxic and ADCC activities of phagocytes. Cultured witha Lenolate % 51Cr releasec treatmentb Cytotoxicity ADCC PMN -5.0 ±2.0 NTd PMN + 4.9±1.2 NT PMN+Antiserum -NT 15.8±0.7 PMN+Antiserum + NT 28.7±0.6 PMN+NMS -NT 4.5 ±1.0 PMN+NMS + NT 14.0±0.7 Alveolar MØ -3.2±0.4 NT Alveolar MØ + 3.6±1.2 NT Alveolar MØ+Antiserum -NT 3.0±0.8 Alveolar MØ+Antiserum + NT 3.6±0.4 Alveolar MØ+NMS -NT 2.5±0.5 Alveolar MØ+NMS + NT 2.3±0.8 a) PMN or alveolar MØ (5.0x105 cells/culture) were added to MDCK cell cultures (1.0x104 cells/culture) infected with influenza virus at an m.o.i. of 1.0, 6 hours after infection. Anti-influenza antiserum or normal mouse serum (NMS) (1:200) prepared from BALB/c mice was added to the culture concomitantly with the addition of phagocytes.b) BALB/c mice were injected per os with Lenolate (500 mg/kg) 7 days before obtaining the phagocytes.c) 51Cr released by phagocytes or phagocytes plus sera was measured 12 h after the addition of phagocytes. Resultswere expressed as the mean ±SD in triplicate cultures.d) Not tested fected cells was potentiated by the treatment with Lenolate (Table 3, p.11). These facts might imply that the cytolytic mechanism of PMN in the absence of antibody was different from that in the ADCC response. With regard to the protection in the late phase of infection, treatment of normal or CY-pretreated mice with Lenolate could lead to enhancement in the virus elimination from the lungs (Figure 2, p.7). Furthermore, Lenolate-treatment could enhance the production of neutralizing antibody in both normal and CY-pretreated mice (Figure 4, p.9). There was already evidence that anti-body played a protective role from the transient plateau to late phases of influenza virus infection.25 In a recent study we demonstrated that antibody response, depressed by anticancer drugs, could be restored by the treatment with Lenolate (unpublished data). Therefore, it appears that the effects of Lenolate on PMN and anti-body production in normal or immuno-compromised mice contribute to the potentiation of the mechanisms involved in virus elimination in the late phase of the intranasal infection. References 1. Mitsuyama M, Takeya K, Nomoto K, Shimotori S: Three phases of phagocyte contribution to resistance against Listeria monocytogenes. J Gen Microbiol, 1978; 106: 165-171. 2. Tatsukawa K, Mitsuyama M, Takeya K, Nomoto K: Differing contribution of polymorphonuclear cells and macrophages to protection of mice against Listeria monocytogenes and Pseu-domonas aeruginosa. J Gen Microbiol, 1978; 115: 161-166. 3. Tsuru S, Nomoto K, Mitsuyama M, Zinnaka Y, Takeya K: Importance of polymorphonuclear leukocytes in protection of mice against Escherichia coli. J Gen Microbiol. 1981; 122: 335- 338.
leukocytes and of alveolar macrophages to protection during the early phase of intranasal infection. J Gen Virol, 1987; 68: 425-432.
11.Stein RS, Breaman RN, Ali MY, Hansen R, Jenkins DD, Jummean HG: Lithium carbonate attenuation of chemotherapy-induced neutropenia. New Eng J Med, 1977; 297: 430-431. 12.Berlin, BS: Sparing effect of X-rays for mice inoculated intranasally with egg-adapted influenza virus, CAM strain. Proc Soc Exp Biol Med, 1964; 177: 864-869. 13.Werner GT, Jentzsch U, Metzger E, Simon J: 1980. Studies on poxvirus infections in irradiated animals. Arch Virol, 1980; 64: 247-256. 14.Hurd J, Heath RB: Effect of cyclophosphamide on infections in mice caused by virulent and avirulent strains of influenza virus. Infect Immun, 1975; 11: 886-889. 15.Robinson TWE, Cureton RJR, Heath RB: The effect of cyclophosphamide on sendai virus infection of mice. J Med Microbiol, 1969; 2: 137-145. 16.Singer SH, Noguchi P, Kirschstein RL: Respiratory disease in cyclophosphamide-treated mice. II. Decrease virulence of PR 8 Influenza virus. Infect Immun, 1972; 5: 957-960. 17.Walker M: Olive leaf extract. New York: Kensington Publishing Corp. 1997. 1-198. 18. Habu S, Fukui H, Shimamura K, Kasai M, Nagai Y, Okumura K, Tamaoki N: In vivo effects of anti-asialo GM1. I. Reduction of NK activity and enhancement of transplanted tumor growth in nude mice. J Immunol, 1981; 127: 34-38. 19.Luckcher AE, Braciale VL, Braciale T: In vivo effector function of influenza virus-specific cytotoxic T lymphocyte clones is highly specific. J Exp Med, 1984; 160: 814-826. 20. Neumann C, Sorg C: Immune interferon I. Production by lymphokine-activated murine macrophages. Eur J Immunol, 1977; 7: 719-725. 21.Bukowski JF, Woda BA, Habu S, Okumura K, Welsh RM: Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J Immunol, 131: 1983; 1531-1538. 22.Habu S, Akamatsu K, Tamaoki N, Okumura K: In vivo significance of NK cell on resistance against virus (HSV-1) infections in mice. J Immunol, 1984; 133: 2743-2747. 23.Rodgers B, Mims CA: Interaction of influenza virus with mouse macrophages. Infect Immun, 1981; 31: 751-757. 24.Rodgers B, Mims CA: Role of macrophage activation and interferon in the resistance of alveolar macrophages from infected mice to influenza virus. Infect Immun, 1982; 36: 1154-1159. 25.Iwasaki T, Nozima T: Defense mechanisms against primary Influenza virus infection in mice. I. The roles of interferon and neutralizing antibodies and thymus dependence of interferon and antibody production. J Immunol, 118: 256-263, 1977. |

This website is managed by Riordan Clinic
A Non-profit 501(c)(3) Medical, Research and Educational Organization
3100 North Hillside Avenue, Wichita, KS 67219 USA
Phone: 316-682-3100; Fax: 316-682-5054
© (Riordan Clinic) 2004 - 2024c
Information on Orthomolecular.org is provided for educational purposes only. It is not intended as medical advice.
Consult your orthomolecular health care professional for individual guidance on specific health problems.
 Download The Full Text Article in (PDF)
Download The Full Text Article in (PDF)